Evaporating solvents away from your product is, and I’m sure always will be, a standard part of pretty much any workup procedure. Like most labs I have a rotary evaporator, but unlike most labs mine sits in the corner gathering dust, because I use a different thing entirely.
Before Vapourtec manufactured flow chemistry platforms, we (the royal we, I wasn’t in the company then) developed the V10 evaporator, now owned and sold elsewhere. But, I have the original prototype in my lab here at Vapourtec. It’s not showroom standard, let’s get that out of the way from the start, but for lab scale syntheses, boy is it good. Much of my work is in the 10-20 ml scale and collected into 20 ml synthesis vials. These fit directly onto the V10 and the solvent is pulled away, leaving your now dry and solvent-free sample in a sample vial and that’s it. Either it goes into the dessicator for a bit to make sure it’s totally dry or for samples that were never wet in the first place, goes straight in the post to the NMR lab. No hassle, no having to bend a spatula to reach that really awkward bit just below the neck of the round-bottomed flask, no having to worry about the person before you bumping their sample up inside the rotovap, ready to contaminate your own (though I’m the only person in the lab, so I wouldn’t really have anyone else to blame). Its handy for drying down larger volumes too, there is a piece of tubing that I can connect to a pump, and continuously add solution while the evaporation is still running; pump slower than the rate of evaporation and there’s no risk of bumping.
I do have the rotovap on standby if the V10 had a problem, but in the 2 years I’ve been at Vapourtec it hasn’t (often) missed a beat.
Sometimes only a really strong base will do, and at times butyllithium (BuLi) is the only real contender. BuLi is used a lot in batch processes but, because of its often exothermic behaviour, it is an ideal candidate for flow chemistry – Especially with a reactor that can cool to -70 °C and has pre-cooling on all of the reagent lines coming in.
The most common issues with organolithium reagents are how quickly they become deactivated if they get exposed to moisture, and that some can flame on contact with air. I’ve used n-BuLi quite a lot in flow, and I have to say, it makes it much easier. I suppose it is important to say that n-BuLi is really rather tame compared to its more energetic brother t-BuLi, but the principle is the same. The major benefit of handling it in flow, is that the BuLi doesn’t actually come into contact with air at any point, preventing it from being deactivated.
Or catching fire. Which is usually an advantage.
We use long needles on our aspiration lines (called the organometallic kit) to pierce the seal of the BuLi bottle and pump it directly, without having to try and dispense it first. This is especially easy with the V-3 pumps of the E-Series or R2S. I can also put an inert supply over the bottle to make sure it stays dry. The inert gas manifold on either system makes this simple, and unlike the traditional balloon, the supply doesn’t run out.
Now, you might think that running a solution that can very easily form solids if there is moisture present is a bit risky in a flow reactor, but to be honest the way to solve the problem is exactly the same as you would use if preparing for a batch reaction – dry the system first. It’s quite easy in flow, take an anhydrous solvent, preferably one that is quite hygroscopic (methanol works well) and flush the whole system with it, then I usually flush this away with anhydrous hexane, but THF works perfectly as well. Test everything is dry by pumping a small slug of BuLi – if it fizzes when it comes out of the end you know it was dry…
“Fatal if inhaled”. There. That got your attention.
It certainly got mine! I was preparing the COSHH assessment for a recent application note when I saw that alarming statement, which on the SDS is then followed by the rather helpful advice “do not breathe fume/vapour”, just in case you hadn’t fully understood the concept of fatal if inhaled…
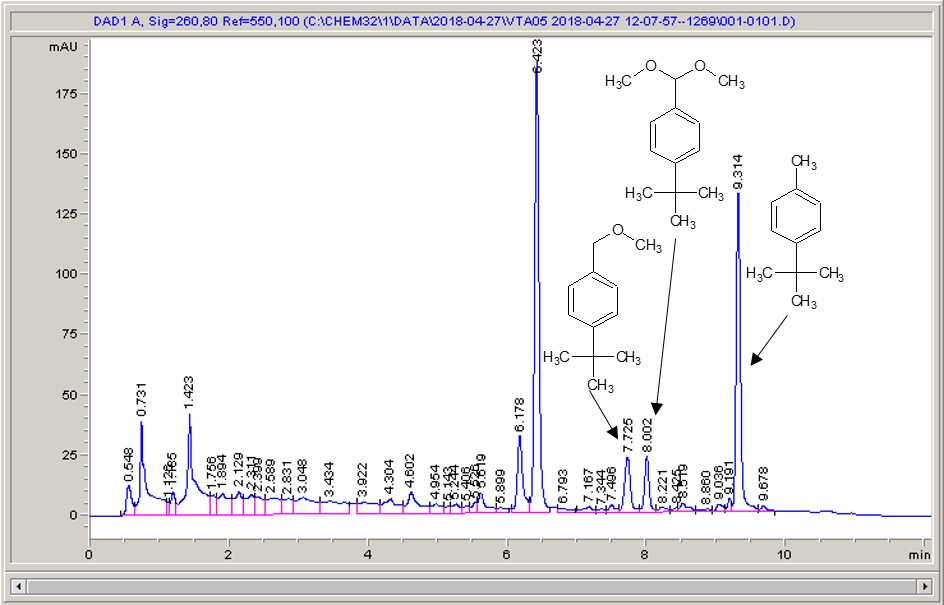
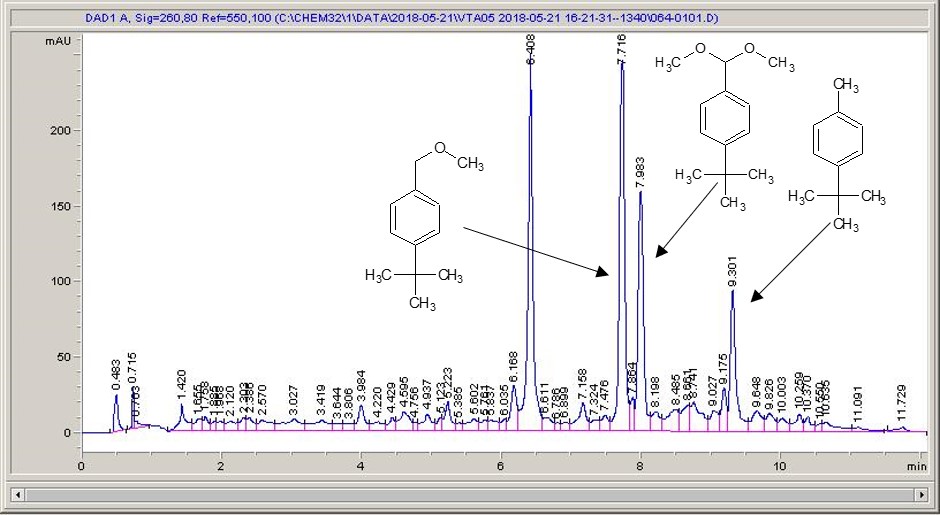
Happily, things like this are made a little simpler with flow chemistry. The plan was to carry out the photochemical bromination of a range of different substituted toluenes, using molecular bromine in solution, and see how easy it would be to increase the throughput of the reaction. As the bromine does present a fairly significant hazard, Dr Roger Moses joined me in the lab to share his experience and provide Devon vs Cornwall related banter (cream and then jam, obviously).
With the bromine in solution, which when I think about it I ought to have made using an SF-10 to deliver the bromine instead of using a Pasteur pipette, working with it in flow was very straightforward. It was very pleasing to see the orange-brown reagent mixture going into the reactor and colourless product coming out of the other end, although thick gloves were needed for everything, as the HBr generated in a stoichiometric amount, can fume off of the product vial. A little wash with some thiosulfate solution to pick off any bromine that might still be lurking about, and then onto the HPLC to get a feel for what’s going on. As far as boosting throughput goes, this really was about as easy as it gets; the UV-150 is very efficient, so even with residence times of just a minute we were seeing conversions of 80% or so. We quickly got up to around 15 grams per hour, and then chickened out pushing any further – the benzylic bromides are well known for their lachrymatory properties, and cleaning up a bunch of essentially tear gas didn’t seem like a fun way to spend the afternoon.
Perhaps unsurprisingly, a key aspect of electrochemistry, is the electricity.
I know, ground-breaking stuff! As I’ve discussed a lot recently, the Ion electrochemical reactor has been developed with a lot of different features, like temperature control and operation under pressure, but I realised I’ve never actually mentioned the important bit – the electrics.
The Ion will have its own power supply, but up to this point I’ve been working with an early prototype. A really important feature has been that I can set the desired current and monitor the change in the voltage, or the other way around. Mostly I have been working in a current limited mode, and been adjusting the concentration of my electrolyte to achieve the voltage that I need – this is quite important, because if you have too high a voltage your reagents or products can just be pulled apart and you get ions all over the place. Not enough voltage, and nothing happens.
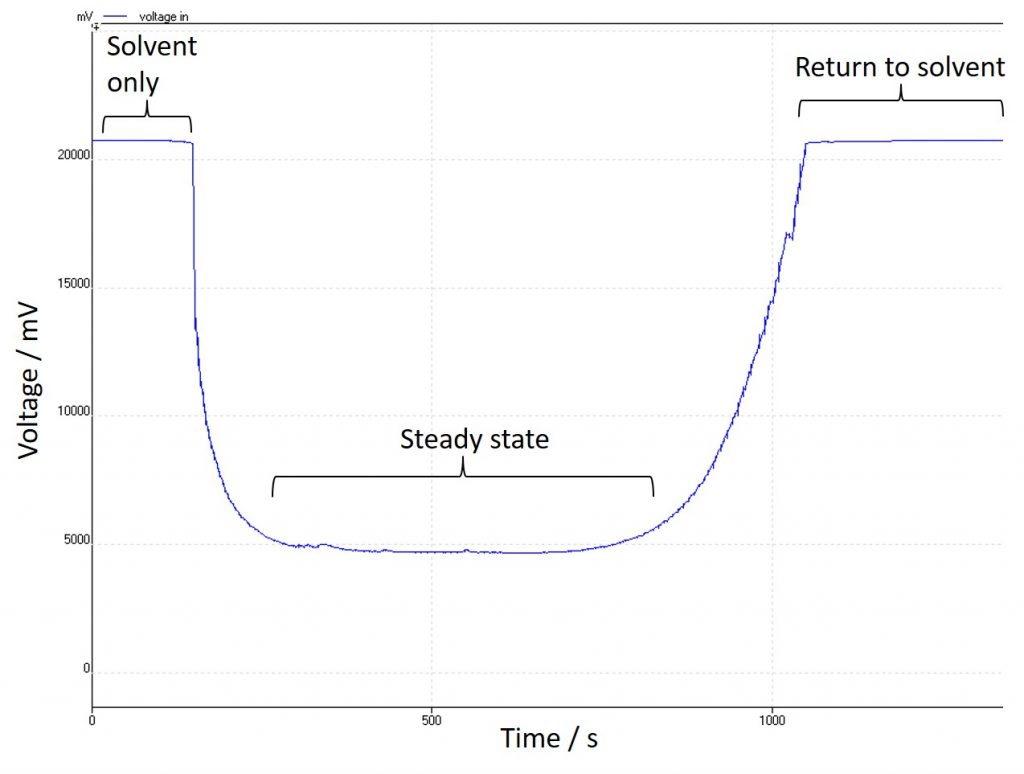
Another handy thing about fixing one parameter and allowing the other to vary is that I can monitor that variation with time – in this case I have been using an external logging device to watch changes in the voltage, but the Ion’s own power supply will come with that function built-in. This is useful for a couple of reasons: firstly, it makes it very clear when the substrate has made it into the reactor. The system solvent does not contain any electrolyte, and so the conductivity is poor, and stays constant. As the substrate enters the reactor the conductivity increases and so the voltage (in my case) needed to achieve the current reduces:
A successful electrochemical reaction
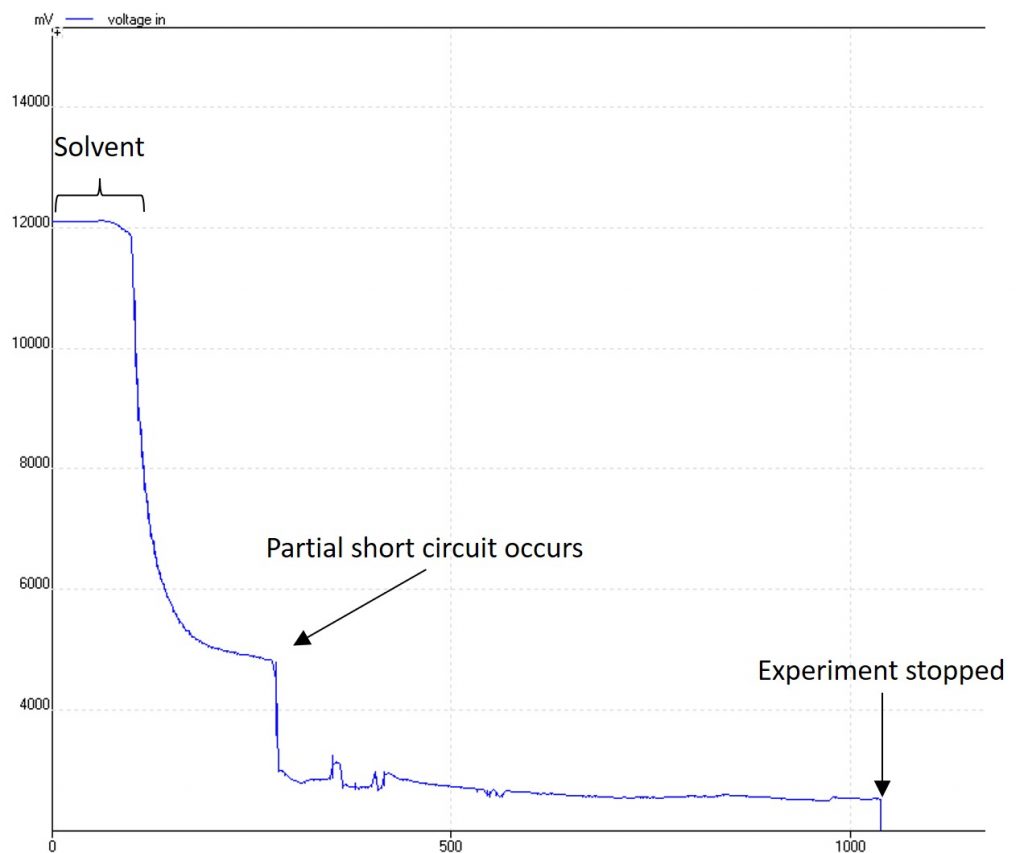
It makes it clear when the reagent is in the reactor which is important for collecting the steady state for analysis. It can also make it very clear when something has gone wrong:
A reaction where a partial short-circuit has occurred
in this image, a build-up of degradation products caused a small short-circuit in an early prototype. The good news is that tracking the voltage with time enabled me to see this right away and stop the experiment, rather than keep running an expensive starting material. The design has also been improved to prevent short circuits like this from happening.
Recently, much of my work has been focussed on using the Vapourtec Ion electrochemical reactor; one of the key features I’ve been investigating is control of temperature. The Ion can be heated well above ambient and cooled to below zero (as we’re still before the official release, I can’t tell you the absolute values), which opens up a vast chemical reaction space to explore. A quick search of the literature, which is already pretty light on electrochemical syntheses, shows that the vast majority of electrochemical reactions are carried out at room temperature; and there are very few examples of cooling an electrochemical reaction.
I suppose the most immediate question is “does temperature have any effect on an electrochemical reaction?” to which I can direct you to a couple of very interesting publications: A group at Pfizer published in 2017 that by incorporating electrochemistry into a nickel-catalysed cross-coupling, they were able to optimise the reaction with much greater control and achieve better conversions and selectivities than with the analogous activated metal powder reactions. The key point here is that the reaction is a thermally mediated coupling, taking place at 65 degrees, but the electrochemistry is used to help control and direct the outcome. A second paper earlier this year from Keio University, Japan, describes a palladium-catalysed iodination performed at 90 degrees. Addition of electrochemical oxidation improved the rate of iodination at hindered positions, and enabled the researchers to direct the reaction to one product over another. Exciting stuff, and both need the ability to heat the electrochemical reactor.
I think there are a number of other phenomena that are temperature dependent to consider: molecules adsorbing to electrode surfaces, followed by ions desorbing; changes in viscosity affecting rates of ion diffusion; the potential for non-electrochemical side reactions – all are worth considering and most probably influenced by the reaction temperature. Certainly in light of the recent literature, combining electrochemistry with thermal processes offers fascinating potential for directing the outcome of reactions.
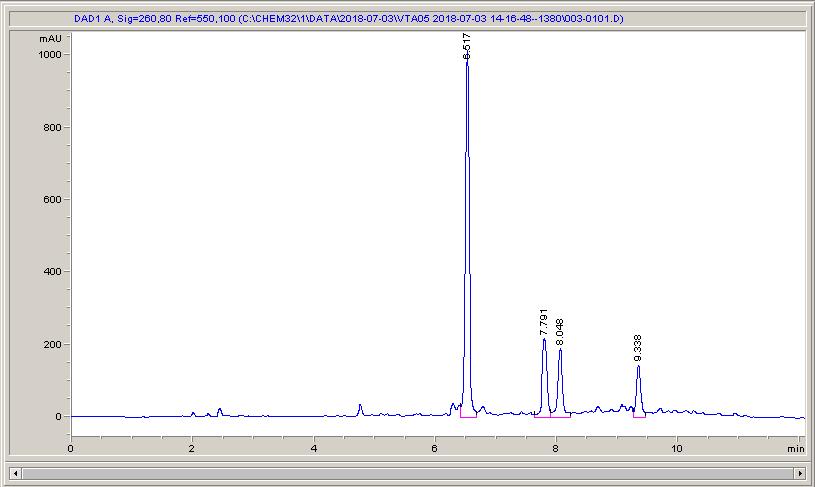
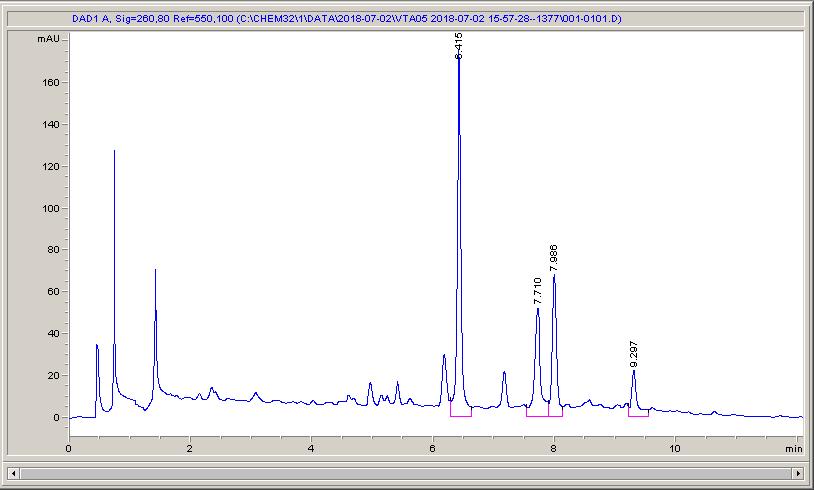
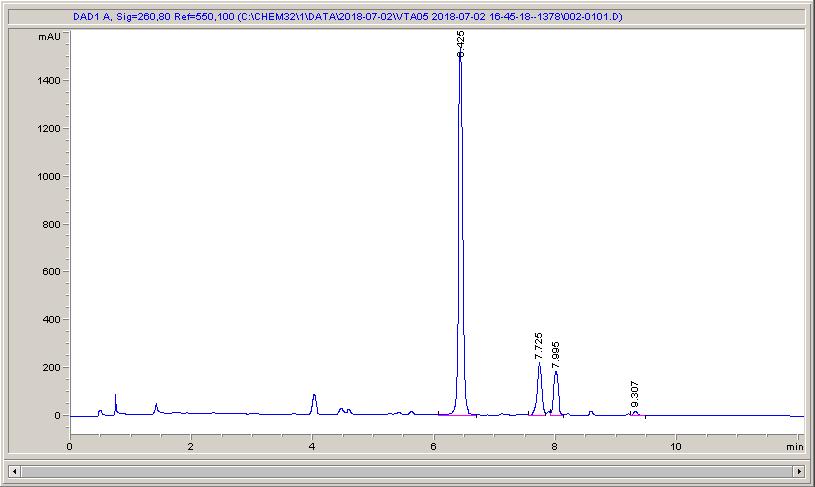
Below are some HPLC traces for my model reaction – the methylation of 4-t-butyl toluene that I’ve written about before. The key point with these experiments is that methanol (reagent and solvent) is taken well beyond its boiling point in the high temperature example. This is only possible because the Ion has been designed to operate at pressure, specifically to allow super-heating of the solvent.
Looking at the results below (I’ve discussed elsewhere that the largest peak, due to an aldehyde, has a much stronger chromophore than the other materials – I have adjusted for this in my integration which is why the apparent peak areas don’t quite much the percentages), the influence of temperature is marginal for this reaction under these conditions.
The model reaction at low temperature – 79% conversion, with 16% reacted over to the aldehydeThe model reaction at low temperature – 84% conversion, and 10% of the products have reacted on to the aldehydeThe reaction at high temperature – full conversion, but 25% of the aldehyde
What I think is most exciting is the limited information in the literature regarding the effect of temperature on these reactions.
Pass me my pith helmet – there’s some exploring to do!
With every R-Series purchased, Vapourtec provides training on-site after the installation. I quite often carry out training and I am always careful to be thorough, even laying traps for the users between experiments to see if they have learned or not. It’s always well received, but then I leave and after a few months, things can be forgotten or new users join the group and aside from the manual, that experience can be lost.
Enter YouTube. It’s the ideal platform to show some videos of an experienced user (which I humbly have to tell you, is me) going through the process of preparing and running a reaction, so that our users have something to refer to if they can’t quite remember. It’s a great idea and has been really well received, but recording the videos can be quite the experience. A professional video maker, Jim – from Green Spark Productions, arrives with a car load of cameras, lights, microphones, and my lab becomes a film studio. I always make an outline of the video, with key points that I need to cover at each stage, and I keep it nearby to refer to. But, to be honest, there are a lot of bloopers. In one particular video in which I am carrying a nanoparticle synthesis I have to say “iron tetrafluoroborate hexahydrate and 1,2,4-triazole” (which are the reagents for this particular synthesis). Needless to say, it took quite a few goes, and when I finally did say it, I had totally forgotten what I was supposed to say next. Not to mention dropping things…
Overall, it’s a great experience though. I’ve really enjoyed making the series so far and the benefits to our customers and distributors around the world are clear. Already (at the time of writing) one of the videos, a Suzuki coupling, has had just over 1000 views! People at conferences have even started mentioning them to me; it’s not easy being a super star, but I manage.
Electrochemistry at Vapourtec has come a long way in just a few short months! Our electrochemical reactor, now known as the Ion, can now operate under a wide range of temperatures, pressures, and using a variety of different electrode materials.
I’ve admitted before that my experience with electrochemistry is limited, and looking through the literature I feel I would be forgiven if I thought that electrode choice didn’t make a difference; especially when using materials such as carbon – I don’t mean electrode pairs, which of course will make a difference as different metals/conductors have different surface potentials.
Throughout the literature, graphite is often called upon to act as an electrode and, more recently, carbon-loaded polymers such as PVDF. The Ion has been designed to take a wide range of electrodes, and as carbon is available in so many different incarnations I thought it would be interesting to trial a bunch of different carbon electrodes under the same electrochemical conditions to see if it makes any difference.
It does.
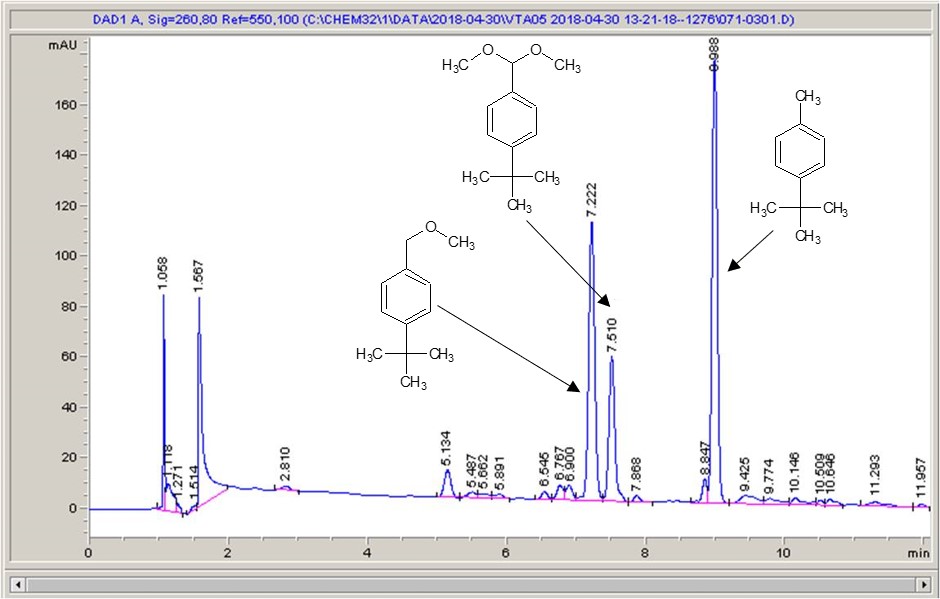
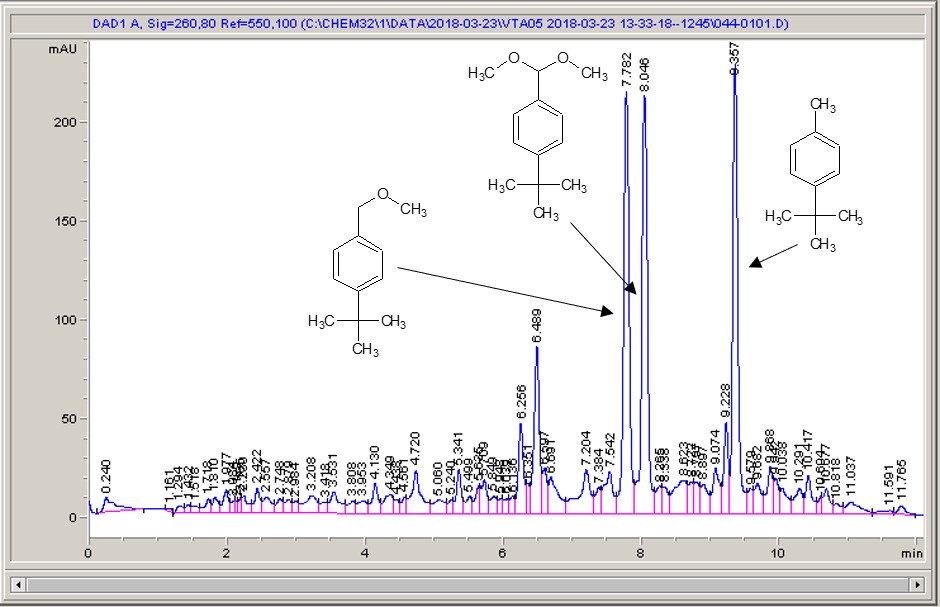
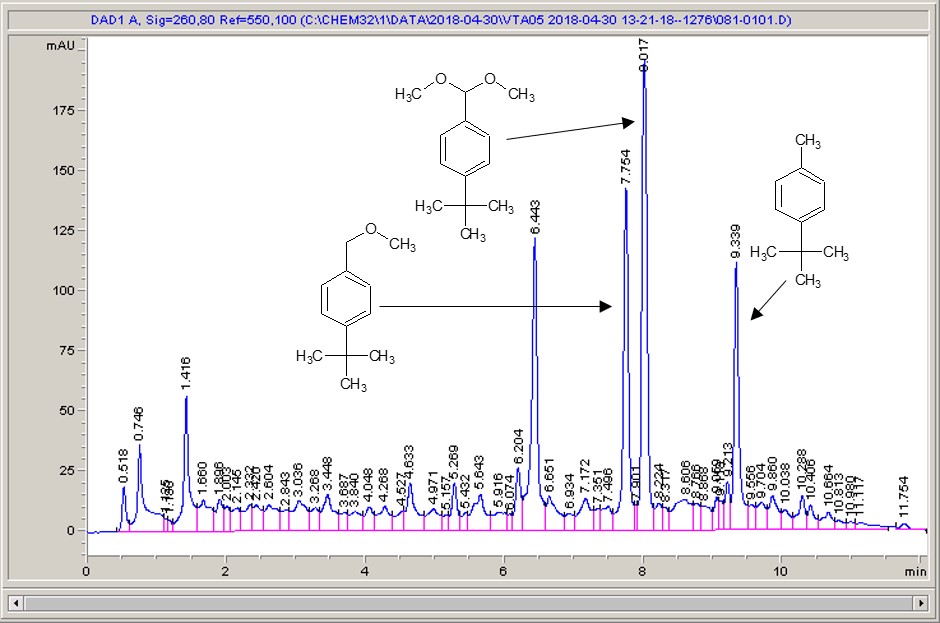
Take a look below; the chemistry is a very commonly studied model reaction, oxidising 4-tertbutyl toluene with methanol to the methyl ether, then the acetal (the aldehyde can crop up as well (see the peak at around 6.4 mins?), but after extensive investigation I found that the aldehyde chromophore is 10 x more strongly absorbing than the toluene, so it’s never quite as bad as it looks).
Carbon electrode material 1Carbon electrode material 2Carbon electrode material 3Carbon electrode material 4Carbon electrode material 5
What I see right away is a big difference between each of the different materials. Carbon-loaded PVDF (material number 4) has resulted in a rather poor conversion with a significant amount of the aldehyde (even accounting for the stronger chromophore), whereas some of the other materials have quite high conversions, and some show the start of interesting selectivities. The baseline noise changes considerably between electrodes as well. Each measurement is preceded by a blank solvent run for comparison, and they always come through clean.
In early 2017, I spent two weeks working off site with a collaborator, New Path Molecular Research, helping to develop what would become Vapourtec’s peptide synthesis platform. Crikey what a project it was, talk about fascinating! The overall concept is deceptively simple, as all good concepts are really; you start with some form of solid support, typically some derivative of a Merryfield resin, attach the first amino acid of your peptide sequence to it, and then just keep adding on the right amino acids until your sequence is finished. Chemically detach the end from the support and hey presto! Your nice and clean peptide is ready to use.
The technical challenge was a little more complex, but flow chemistry is ideal for this procedure; batchwise solid-phase peptide synthesis (SPPS) is, frankly, fiddly. Every amino acid you want to add, you first have to deprotect the terminal amino acid already in the sequence, wash and dry. Add the next amino acid and give it sufficient time to react, wash and dry. Deprotect again, wash and dry and so it goes on until your sequence is finished. Each step can take hours in total, and three or four couplings a day is about the best you can hope for. The advantage that flow brings immediately is that the resin is being constantly washed, merely by virtue of being in flow. So that step of the process is now redundant. Also, in flow we can easily pre-activate our amino acids, usually to an ester, so the coupling is fast; this can be done in batch too, but the activated species must be used quickly, otherwise it can racemise or decompose. In flow we can pre-activate the amino acid quickly and pass it immediately over the deprotected sequence on the resin.
The new platform is complex, it’s probably the most advanced Vapourtec configuration I’ve worked on but with that complexity comes some impressive capability (which can be read about in detail here). Automated liquid handlers let you dial in the sequence you need, and inline spectroscopic and physical measurements allow real-time monitoring of the efficiency of each coupling, and if a coupling hasn’t worked properly? Run it again. As long as the newly added amino acid is not deprotected first, you can only build the sequence that you want.
I love a conference, and Achema really is one of the biggest around! Once every 3 years, specialists from around the world gather to showcase chemical equipment and technology. The most recent took place a couple of weeks ago in Frankfurt, and I was lucky enough to be there with Vapourtec.
Conferences are a fantastic opportunity to meet people in the flow chemistry community. It’s always great to talk to other vendors, catch-up with existing customers and hopefully meet some new ones. I also had the chance to present some of the application work that I have done, which is always an exciting opportunity; I spoke in detail for the first time about Vapourtec’s collaboration with New Path Molecular in using an R-Series with a newly developed reactor that, when combined, makes a platform capable of automated peptide synthesis (which will be featured in an upcoming post). I’m really proud to have been part of this project and how the development of new technology has been able to tackle some of the chemical challenges we faced – a theme I have followed since starting my PhD with self-optimising reactors. The talk was well received and resulted in quite a lot of people visiting me at the stand to discuss the chemistry further, which is nice (my slides are here, if you’re interested).
Unique to this conference, all of the flow chemistry vendors allied together and created the Flow Chemistry Pavilion: a focussed concentration of all of the vendors together. I’ll admit I was a little sceptical to start with, we have competitors and they were in close proximity, but the reality is that it worked very well; people with any interest in flow were coming to the pavilion and because everyone was there, we all saw much higher footfall. It also led to a great feeling of community, as all of the vendors started to direct people to the most appropriate supplier for their application: “Need a scale-up option? You need to go and see so and so, just over there.” “Inline separation you say? Ah, well that chap there is the expert in that.”
Goodness pressure is important in continuous flow. In fact, one of the major advantages of flow vs batch is how easy it is to apply pressure. The main benefit of pressure for a homogeneous liquid reaction is that the solvent can be heated above its standard boiling point, allowing you to give the kinetics of your reaction a bit of a kick up the proverbial and reduce the time needed inside the reactor, and thus, increase your throughput. It does go without saying though that this approach only applies to chemistry robust enough to take it, and those who are at the final stage of a synthesis of a large natural product, probably need to take things more gently.
That said, there are other reasons that pressure is important – we can discuss thermodynamic, physical chemistry functions like the Gibbs volume of activation if you like, but I think before we need to think too hard about that, pressure has more immediate benefits. Preventing gas evolution from reducing residence times is key to maintaining good conversions, and pressure is vital in gas/liquid reactions for making sure the liquid is saturated with the gas for the reaction to take place.
But, there is a problem. Solids, particulates, precipitates of any kind can cause a real problem with needle and seat-type back-pressure regulators, causing inconsistent pressure management at best, and blockages at worst. Enter the peristaltic BPR. The SF-10 pump uses the Vapourtec V-3 pumps and can run as a pressure regulator. Now, the maximum pressure is 10 bar, but that’s more than enough to superheat most solvents quite a long way and the continuous release of pressure, instead of the “open-closed” binary state of a needle BPR, allows for much smoother, more consistent pressure control. The real piece de resistance though is that the SF-10 can handle solids. Not just the odd crystalline precipitate either, I’ve used these pumps to control the pressure of slurries loaded up to 100 mg/ml without any trouble and now it’s my go-to method of controlling pressure, simply because I know that it will just keep working even if I haven’t got my down-stream solubility quite right (which I never have, by the way).